AlphaGenome Just ‘Solved’ 98% of the Human Genome – Here’s Why That’s Both Brilliant and Bullshit
DeepMind just dropped what they’re calling a “foundational step” toward decoding life’s operating system. AlphaGenome, published in Nature on January 28, can process one million base pairs of DNA in a single pass and predict how mutations affect everything from gene expression to chromatin folding. The model matches or beats 25 of 26 existing benchmarks for variant effect prediction. Impressive? Absolutely. But the gap between press release triumphalism and bench scientist skepticism reveals a familiar pattern in AI research: when the training data is messy, the “ground truth” is disputed, and the compute requirements demand a small power plant, even breakthroughs come with asterisks the size of chromosomes.
The Technical Marvel: A Million Letters at Once
Let’s give credit where it’s due. AlphaGenome’s architecture is genuinely clever. The model uses a U-Net-inspired backbone with convolutional layers for local patterns and transformers for long-range dependencies, processing 1 Mb sequences across eight interconnected TPUv3 devices. That’s not a weekend Kaggle project, that’s “industrial scale” engineering, as computational biologist Mark Gerstein noted.
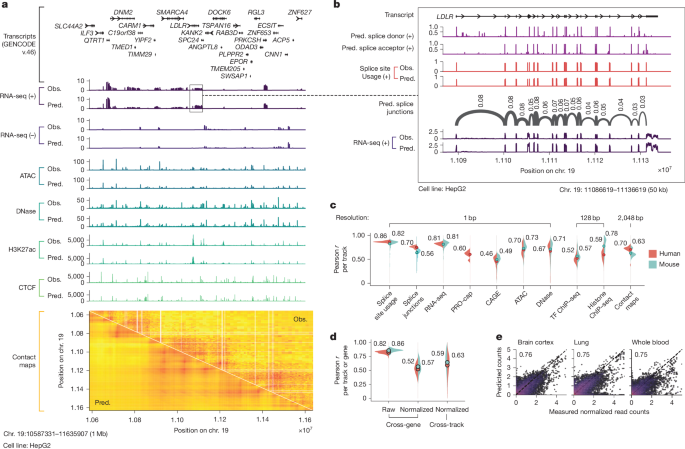
The model predicts 5,930 human genome tracks across 11 modalities: RNA-seq coverage, splice site usage, chromatin accessibility (ATAC-seq/DNase-seq), histone modifications, transcription factor binding, and even 3D contact maps. It does this at base-pair resolution for many outputs, a feat previous models couldn’t manage without trade-offs.
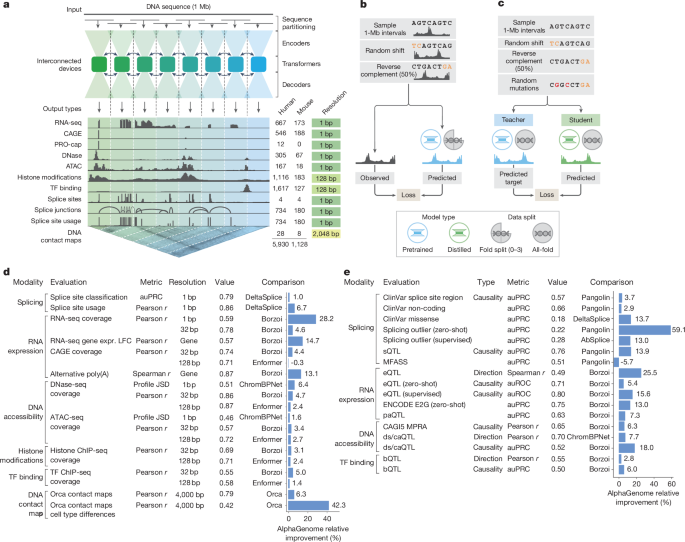
The key innovation is the two-stage training process. First, a pretraining phase uses 4-fold cross-validation on experimental data to create both fold-specific and “all-fold” teacher models. Then comes distillation: a single student model learns to reproduce the ensemble’s predictions using randomly mutated input sequences. This makes the final model both robust and efficient, variant effect predictions take <1 second on an H100 GPU, compared to the cost of ensembling multiple independent models.
For the TAL1 oncogene, a gene that causes T-cell acute lymphoblastic leukemia when misregulated by mutations 8,000 bases away, AlphaGenome accurately predicted which mutations would create a “super-enhancer” that permanently activates the gene. Marc Mansour, a hematologist at UCL who discovered these mutations experimentally, called it “mind-blowing.” That’s not nothing.
The “Dark Genome” Myth: When Marketing Meets Biology
Here’s where the bullshit detector starts beeping. The press release narrative frames the 98% of non-coding DNA as the “dark genome”, a mysterious void that AlphaGenome finally illuminates. It’s a compelling story, AI conquering biology’s final frontier, but it’s scientifically misleading.
The non-coding genome isn’t “dark matter.” Scientists have been studying regulatory elements, enhancers, promoters, and insulators for decades. The ENCODE project spent $400 million mapping functional elements. The problem isn’t that we didn’t know these elements existed, it’s that predicting their exact function from sequence alone is computationally brutal and biologically nuanced.
As Steven Salzberg, a computational biologist at Johns Hopkins, bluntly put it: “I see no value in them at all right now. I think there are a lot of smart people wasting their time.” His critique cuts deeper than typical academic sniping. The community still can’t agree on a gold-standard set of splice sites after 25 years. If the training data itself is contested, what exactly is the model learning?
This isn’t just theoretical nitpicking. AlphaGenome’s training data comes from experimental assays that are themselves noisy, cell-type-specific, and often contradictory. When you train on data derived from different labs, protocols, and cell lines, you’re not learning “the regulatory code”, you’re learning a weighted average of experimental artifacts.
The Benchmarking Paradox: Winning the Game vs. Solving the Problem
AlphaGenome’s performance numbers look stellar: 25 of 26 variant effect predictions match or exceed state-of-the-art. But this is where we need to talk about critiques of AI benchmarking reliability and model robustness. The AI community has been playing a rigged game where models ace specific tests but collapse under distribution shift.
Consider the eQTL prediction task. AlphaGenome achieves a Spearman ρ of 0.49 for effect size prediction, up from Borzoi’s 0.39. For sign prediction (will expression go up or down?), it hits 0.80 auROC versus 0.75. These are modest improvements, not paradigm shifts. And performance degrades significantly for variants more than 35 kb from the transcription start site, the exact region where long-range models should theoretically excel.
The model also struggles with personal genomes. As David Kelley from Calico Life Sciences noted, “AlphaGenome predicts on one standard human genome”, but real patients have millions of variants interacting in complex ways. This is the difference between predicting weather in a controlled lab environment and forecasting actual climate patterns.
Even the TAL1 success story comes with caveats. AlphaGenome’s predictions are most confident for variants within 10-20 kb of the gene. The truly distal enhancers, those operating 100+ kb away, remain poorly predicted. It’s like claiming you’ve mastered long-range forecasting because you can predict tomorrow’s weather.
The Compute Reality Check: Who Can Actually Use This?
Here’s the dirty secret: that 1 Mb context window requires serious hardware. The paper mentions “sequence parallelism across eight interconnected tensor processing unit (v3) devices.” That’s not a laptop setup, that’s a Google-scale infrastructure requirement.
This raises questions about Google’s extreme measures for AI compute scalability. When your model’s inference requirements are so intensive that they factor into corporate energy planning, you’ve created a tool that only a handful of institutions can actually deploy at scale.
Yes, DeepMind provides an API and the distilled student model runs on a single H100. But the full power, the ability to retrain on new cell types or species, is locked behind a compute wall. This creates a two-tiered system: API users get a black box, while only Google-scale labs can truly experiment with the architecture.
The Clinical Translation Gap: From Bench to Bedside
Natasha Latysheva, a research engineer at DeepMind, envisions AlphaGenome accelerating drug target discovery. And clinicians like Marc Mansour are already using it to prioritize cancer mutations. But Katherine Pollard, a data scientist at Gladstone Institutes, offers a crucial reality check: “It is a much, much harder problem, and yet that’s the problem we need to solve if we want to use a model like this for health care.”
The model predicts molecular effects, not disease phenotypes. It doesn’t account for gene-gene interactions, environmental factors, or developmental timing. A mutation that activates an oncogene in a stem cell might be harmless in a differentiated tissue. AlphaGenome can’t yet model these contextual dependencies.
This is where we need to be honest about AI-driven breakthroughs in cancer research and biological discovery. The gap between predicting a variant’s molecular effect and understanding its clinical impact is enormous. AlphaGenome can tell you what might happen to gene expression, but not why that matters for a specific patient’s disease trajectory.
The Data Paradox: Google’s Advantage or Albatross?
DeepMind trained AlphaGenome on human and mouse data from public databases, ENCODE, GTEx, 4D Nucleome. This is both a strength and a limitation. On one hand, it’s reproducible science. On the other, it highlights Google’s struggles to leverage massive data for AI dominance.
Unlike protein structures (where AlphaFold had clean, consensus data) or Go (where the rules are unambiguous), genomic regulation is a field where the “rules” are still being written. Having more data doesn’t help if that data is contradictory or incomplete. DeepMind’s industrial-scale compute can’t fix fundamental biological uncertainty.
The model’s performance on the “dark genome” is particularly telling. For Mendelian disease variants, setting a high threshold on AlphaGenome scores enriches for causal variants, but at the cost of low recall. For complex trait GWAS variants, the enrichment is even weaker. The model is better at ruling things out than definitively identifying causal mechanisms.
The Verdict: Powerful Tool, Not a Crystal Ball
AlphaGenome represents real progress. The engineering is impressive. The unified approach to multiple modalities is smart. The TAL1 example shows genuine utility. But it’s not AlphaFold, and it’s not going to win a Nobel Prize, not yet.
The model excels at hypothesis generation and prioritization. It can tell you which variants in a cancer genome are worth testing first, or suggest regulatory elements to target in a synthetic biology project. But it can’t replace experimental validation, and it certainly can’t diagnose patients from whole-genome sequences.
As we navigate this new era of AI-powered biology, we need to avoid the trap of magical thinking. AlphaGenome is a sophisticated statistical model trained on incomplete data. It’s brilliant at interpolating within the bounds of what we’ve already measured, but brittle when extrapolating beyond them.
For researchers, the message is clear: use AlphaGenome as a compass, not a map. Let it guide your experiments, but don’t let it replace your judgment. And for the rest of us? Maintain a healthy skepticism when reading headlines about AI “solving” the genome. The code of life is written in chemistry, context, and chance, three things that no model, however powerful, has yet mastered.
What do you think? Is AlphaGenome a genuine breakthrough or another case of AI hype outpacing reality? Have you experimented with the model? Share your experiences in the comments.
For more on the intersection of AI and biological discovery, check out our coverage of AI-driven breakthroughs in cancer research and biological discovery and why critiques of AI benchmarking reliability and model robustness matter more than ever.